
Project Orbis: A three-year review
By Xcenda

Overview of Project Orbis
Project Orbis is a collaborative program initiated by the United States (US) Food and Drug Administration (FDA) Oncology Center of Excellence (OCE) in 2019 that provides a framework for concurrent submission and review of oncology products among international regulatory partners. The aims of the program are to allow patients faster access to promising cancer drugs and, ultimately, to establish a greater uniformity of cancer treatment standards across the globe. Prior to the launch of the program, collaboration between the FDA OCE and its counterparts in partner countries primarily involved monthly teleconferences to discuss general topics and specific product marketing applications. Through these conferences, it became apparent that the FDA typically received product applications months or even years before other partner countries. Participation in Project Orbis expands the interactions of the FDA and partner regulatory health authorities (Table 1) to include direct collaboration with application review, allowing partners access to the same submission data reviewed by the FDA while maintaining full independence when making regulatory and labeling decisions.
Table 1. Regulatory authorities participating in Project Orbis

The FDA OCE decides on eligibility for the project and coordinates the application selection, review process, and development of a core review document to facilitate discussions among Project Orbis Partners (POPs).
Eligibility
Products eligible for Project Orbis include new chemical or biological entities and supplemental applications for new oncology indications. Applications should represent high-impact oncology drugs with clinically significant data that would generally qualify for FDA priority review because of improvements in safety or efficacy. The FDA can recommend an application for Project Orbis based on a combination of breakthrough designation, impressive results, and unmet need. Sponsors, such as biopharmaceutical companies responsible for commercialization of a new product/indication, may also submit proposals to the FDA Office of Oncologic Diseases’ review to determine suitability for Project Orbis, including topline efficacy and safety results from clinical trials and a global submission plan. Proposals cannot be submitted to other POPs, and inquiries to partners are referred to the FDA.
Pre-submission activities
Once an application is nominated, the FDA review team notifies the OCE global team, who then reaches out to the POPs to determine interest and availability to collaborate on the review. Participation of each POP is contingent on the application meeting any additional requirements specific to the partnering regulatory authority. For example, Health Canada’s participation requires that an application meet its criteria for an expedited pathway (ie, Health Canada Priority Review or Advance Consideration for a Notice of Compliance with Conditions) in addition to meeting the FDA criteria for priority review. The FDA will notify the sponsor company of acceptance into Project Orbis and which POPs have agreed to participate. While sponsors can select a minimum of two POPs (one of which must be the FDA), they are encouraged to submit their application to all interested POPs. Sponsor companies should work with each POP to determine whether pre-submission meetings are required, confirm POP-specific regulatory requirements, and negotiate pre-submission and submission timelines.
The FDA confirms the global submission plan through the US-based sponsor, formally designates participating POPs, and establishes a Project Orbis Working Group (POWG) that includes the FDA and participating POPs for each application. In the case of a US-based sponsor with limited international operations, establishment of a legal entity in the intended POP country, engagement of a third party to act as the applicant, or both may be necessary. There are three types of Project Orbis submission plans, depending on the timelines for submission to the FDA and participating POPs, with varying levels of concurrent review and action (Table 2). POPs determine which type of collaboration is possible for them, based on priorities and workload.
Table 2. Types of Project Orbis submission plans
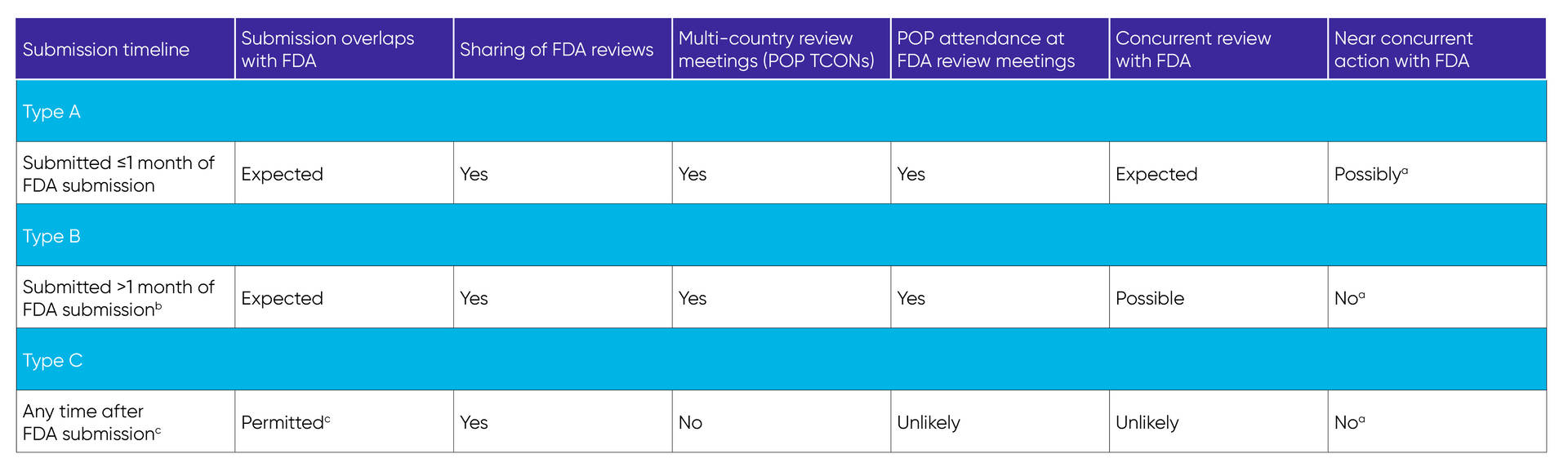
FDA – US Food and Drug Administration; POP – Project Orbis Partner; TCON – teleconference.
aRegulatory action in other jurisdictions is unlikely to occur immediately after FDA action and will follow respective health authority timelines.
bThis is still within a time frame allowing POP participation in review meetings.
cThis is dependent on POP guidelines. Contact specific POP(s) regarding optimal timing for submission of Type C dossier.
Source: Project Orbis Types Table-v2 (Rev. Jan 2023). Accessed February 2, 2023. https://www.fda.gov/media/165101/download
Submission
Product marketing applications (ie, dossiers) must be submitted to each participating regulatory health authority (RHA) using the electronic Common Technical Document format in English, with possible exceptions for the country-specific components of the application, and conform to the respective domestic submission requirements for each RHA. For type A or B Orbis submissions, the use of the Assessment Aid (AAid) document is required. The AAid is divided into three parts:
- Data (concise presentation of objective results provided by the sponsor)
- The Applicant’s Position (interpretation of data completed by the sponsor)
- FDA’s Assessment (assessment of the data and applicant’s position completed by the FDA).
The primary objectives for using the AAid are to (1) focus the FDA review on critical thinking (assessment), and (2) increase review efficiency and consistency, and decrease review time spent on administrative tasks (eg, formatting). The AAid serves as the core document for the POWG discussions during the review process.
Review process
For type A and some type B submissions, the FDA schedules and coordinates a kickoff meeting and several multi-country review teleconferences to discuss aspects of the application (eg, efficacy, safety, clinical pharmacology, risk-benefit assessment). For type A submissions, the kickoff meeting is scheduled before or within 30 days of the FDA application submission to discuss the overall review strategy and review timelines within the POWG. The FDA analyzes the submitted tabulation and analysis datasets to verify clinical trial results and presents key findings for discussion with the POWG during the midcycle meeting. Additional POWG teleconferences include discipline-specific discussions of relevant sections of the AAid. During the review period, each POP conducts its own evaluation of the product marketing application and shares information requests (IRs) for POWG comment and review prior to sending to the sponsor company/local affiliate; the sponsor teams are instructed to send the IR response(s) to all participating RHAs. While the POWG collaborates on evaluation and discussion of the application, each agency retains independence in regulatory decision making to adhere to country-specific laws, regulations, ordinances, and/or policies.
For type B submissions, the entry timeline of the POP with the ongoing FDA review affects the number of multi-country meetings and the possibility of concurrent or overlapping review. For type C submissions, POPs will not have concurrent review or concurrent action with the FDA.
For all Project Orbis submissions, the independent decision making of each RHA may result in differences in the approval or rejection of marketing authorization, the wording of the indications, approval of other labeling content, and post-marketing surveillance requirements.
Project Orbis in practice
From September 2019 to March 17, 2023, there have been 73 FDA approvals through Project Orbis. The number of global approvals include 44 in Australia, 22 in Brazil, 52 in Canada, 12 in Israel, 25 in Singapore, 26 in Switzerland, and 15 in the United Kingdom.
Currently, there are a limited number of reports indicating shortened regulatory timelines for applications reviewed under Project Orbis. During the first year of the program, the median time to approval was similar between the FDA (4.2 months, range 0.9–6.9 months; N=18) and the POPs (4.4 months, range 1.7–6.8 months; N=20).
A recent review of US approvals revealed a shorter median time to approval of 4.2 months for the FDA under Project Orbis compared with 6 months under the FDA priority review or 10 months under standard review. During the first year of Project Orbis, only 17 of 60 marketing applications (28%) were for new molecular entities. It has been suggested that improvement in approval times for submissions reviewed under Project Orbis during the initial pilot program could be related to the high proportion of supplemental applications based on pivotal studies with straightforward study designs. In addition, other FDA-expedited review programs (eg, breakthrough therapy designation, Real-Time Oncology Review) were used in the majority of applications reviewed under Project Orbis during that time, potentially contributing to improved review time frames.
In the first year of Project Orbis, participation for Australia (May 2019–December 2020), 5 evaluations conducted via Project Orbis were completed, with a median time to approval by the TGA of 68 working days (range 35–123 days), compared with a median time of 330 days for approval of new active substances in 2020 under the TGA standard pathway (ie, products not meeting criteria for priority review or provisional approval).
During the first 18 months of United Kingdom participation in Project Orbis (January 1, 2021–June 30, 2022), the MHRA approved 11 of 30 (37%) submissions that received FDA approval. Among those approvals, the average delay in approval between the FDA and the MHRA was 5.7 months compared with 7.8 months between the FDA and the European Medicines Agency. However, the standalone MHRA process is only expected to take 150 days for marketing authorization; therefore, it is unclear whether the UK regulatory timeline is truly expedited with Project Orbis.
Benefits, challenges, and future directions
There are several potential benefits of Project Orbis participation for RHAs, biopharmaceutical industry sponsors, and patients. Both RHAs and product sponsors benefit from the initiative when marketing applications reach POPs sooner than they would otherwise. The collaboration between POPs provides benefit to RHAs through sharing of independent perspectives on regulatory and clinical considerations for oncology marketing applications, with the potential to establish a greater uniformity of new global standards of treatment, leading to the optimal design of pivotal international clinical trials in oncology. Benefits to the biopharmaceutical industry include faster marketing authorization for new products, reduced overall numbers and duplication of IRs, and the potential for using common regulatory dossiers across the different markets. Finally, the collaborative efforts help make potentially life-changing treatments available to patients as quickly as possible while maintaining each RHA’s high standards for assessment of safety and effectiveness.
Challenges for RHAs and product sponsors include additional workload considerations and coordination required throughout the approval process. Sponsors must assess their internal resources against submission timelines and data needs at the global and local affiliate levels to support pre-submission meetings across POPs and to manage simultaneous review of the applications and label negotiations with local POPs. The increased workload for POPs occurs with the addition of multi-country meetings before and during the review cycle. The additional workload and resource requirements for both the sponsor and POPs have been identified as common reasons for non-submission to and nonparticipation of interested RHAs in an Orbis application review. To date, Project Orbis has led to the approval of numerous oncology drugs for patients across the world. Participating RHAs continue to meet quarterly to discuss overall program status and potential process modifications. In the future, other avenues may include overseeing more complex applications (eg, those with companion diagnostic devices) and eventual expansion to cell and gene therapy products.
Sources
- Ankit T, Shrikalp D, Maitreyi Z, et al. Transition of pharmaceutical regulations: the new regulatory era after Brexit. J Pharm Res Int. 2021;33(47A):804-817. doi: 10.9734/JPRI/2021/v33i47A33076
- Australian Government Department of Health and Aged Care Therapeutic Goods Organization. Project Orbis. April 4, 2023. Accessed February 10, 2023. https://www.tga.gov.au/how-we-regulate/supply-therapeutic-good-0/supply-prescription-medicine/application-process/comparable-overseas-regulators/project-orbis
- de Claro RA, Spillman D, Hotaki LT, et al. Project Orbis: global collaborative review program. Clin Cancer Res. 2020;26(24):6412-6416. doi:10.1158/1078-0432.CCR-20-3292
- Eglovitch JS. Top FDA official interested in ‘Project Orbis’ for cell and gene therapies. Regulatory Focus website. February 13, 2023. Accessed March 17, 2023. https://www.raps.org/news-and-articles/news-articles/2023/2/top-fda-official-interested-in-project-orbis-for-c
- FDA. Project Orbis. Accessed January 10, 2023. https://www.fda.gov/about-fda/oncology-center-excellence/project-orbis
- FDA. Project Orbis approvals database. Accessed March 17, 2023. https://view.officeapps.live.com/op/view.aspx?src=https%3A%2F%2Fwww.fda.gov%2Fmedia%2F166295%2Fdownload&wdOrigin=BROWSELINK
- FDA. Project Orbis frequently asked questions. Accessed March 17, 2023. https://www.fda.gov/about-fda/oncology-center-excellence/project-orbis-frequently-asked-questions
- FDA. Project Orbis: Strengthening international collaboration for oncology product reviews, faster patient access to innovative therapies. Press release. December 8, 2020. Accessed February 10, 2023.https://www.fda.gov/news-events/fda-voices/project-orbis-strengthening-international-collaboration-oncology-product-reviews-faster-patient
- Gao YG, Roberts S, Guy A. Maximizing regulatory review efficiency: the evolution of the FDA OCE RTOR pilot. Ther Innov Regul Sci. 2022;56(2):212-219. doi:10.1007/s43441-021-00371-z
- Government of Canada: Health Canada. Project Orbis. Accessed March 17, 2023. https://www.canada.ca/en/health-canada/services/drugs-health-products/international-activities/project-orbis.html
- Kasli I. HPR151 Has Project Orbis facilitated faster access to oncology therapies in Great Britain following 'Brexit'? Value Health. 2022;25(12):S260. doi.org/10.1016/j.jval.2022.09.1280
- Lythgoe MP, Sullivan R. Project Orbis: the UK experience after 1 year. Lancet Oncol. 2022;23(8):978-981. doi:10.1016/S1470-2045(22)00377-1
- Mulchan N, Guy A. Project Orbis: maximizing patient access to new medicines. Regulatory Focus website. February 2021. Accessed March 17, 2023. https://www.raps.org/news-and-articles/news-articles/2021/2/project-orbis-maximizing-patient-access-to-new-med
- Ricci J, Demont E, Machuca-Márquez P, et al. HPR41 Project Orbis to accelerate patient access to innovative cancer therapies: a narrative review. Value Health. 2022;25(12):S240. doi:10.1016/j.jval.2022.09.1173
- Schofield I. Australian regulator on the highs & lows of Project Orbis. Pink Sheet: Pharma Intelligence website. Published May 4, 2022. Accessed February 10, 2023. https://pink.pharmaintelligence.informa.com/PS146124
- Yoffe A, Liu J, Smith G, Chisholm O. Regulatory reform outcomes and accelerated regulatory pathways for new prescription medicines in Australia [published online ahead of print October 21, 2022]. Ther Innov Regul Sci. 2022;1-16. doi:10.1007/s43441-022-00465-2


