
Real-world evidence in United States regulatory decisions
By Xcenda
By:
Aishani Patel, PharmD; Trent McLaughlin, BSc(Pharm), PhD
Updated
November 13, 2020
Real-world evidence (RWE) has garnered increased attention in recent years to advance drug development. The framework for the Food and Drug Administration’s (FDA’s) RWE Program, published in 2018, serves as a roadmap for the inclusion of real-world data (RWD) and RWE in regulatory decisions, including standards on how RWD are defined, collected, and analyzed. This article reviews the use of RWE in United States (US) regulatory decisions after the publication of the FDA’s RWE framework and describes how manufacturers can leverage RWE to optimize their product launch.

Introduction
The Growth of RWE in US Regulatory Decisions
Traditionally, a randomized controlled trial (RCT) is the gold standard for regulatory approval as it enables the understanding of the safety and efficacy of a drug candidate in a controlled environment. However, it has been shown that supplementing RCT data with RWE confers several benefits to the regulatory decision-making process. With RWE, researchers can increase external validity by including populations that are underrepresented in clinical trials. Moreover, RWE provides the ability to overcome the limitations seen in patient recruitment for RCTs. With an increase in novel medicines for rare diseases, the difficulty in patient recruitment for these clinical trials can be alleviated with RWE from external control arms that serve as comparators for studies.
Per the regulatory changes of the 21st Century Cures Act, the FDA is creating an RWE framework that will evaluate the potential use of RWE to support approval of new indications, changes to labeling about product effectiveness, and meeting post-approval study requirements. This includes evaluating the potential use of RWE to support changes to labeling about drug product effectiveness such as adding or modifying an indication (eg, change in dose, dose regimen, or route of administration), adding a new population, or adding comparative effectiveness or safety information.
In 2018, the FDA released a draft RWE framework that outlined the current use of RWE and areas of further program development such as data standards, RWE generated by clinical trials, reporting requirements for various types of studies, gaps in RWD sources, and reliability/relevance determinations. The framework serves as a roadmap for the inclusion of RWD and RWE in regulatory decisions, including standards on how RWD are defined, collected, and analyzed. It also includes guidance on RWE study methodologies and designs that meet regulatory requirements in generating evidence of effectiveness. The goal of the framework is to lead to program implementation, in which manufacturers would have guidance from the FDA as to when it is most appropriate to use RWE, and how to collect and report that RWE to the FDA. The four ways to use RWE to meet the objective are:
- Utilize RWE to capture outcomes and safety data from RCTs.
- Utilize RWE as an external control for single-arm studies.
- Support an efficacy supplement in observational studies.
- Evaluate safety or effectiveness and support regulatory decisions through observational studies.
According to a statement from former FDA Commissioner Scott Gottlieb, “As the breadth and reliability of RWE increases, so do the opportunities for the FDA to make use of this information.” Previously, the FDA has used RWD primarily in its evaluation of safety and in limited circumstances to inform decisions about effectiveness. The FDA’s RWE Program will focus on exploring the potential of RWD/RWE to support regulatory decisions about product effectiveness by using the following three-part approach to evaluate individual applications:

Since the framework’s conception, the FDA has actively been engaging stakeholders, both internally and externally, to ensure a complete understanding of the field and to obtain further input on the RWE program development.
The use of RWE has previously been limited by the FDA to certain circumstances—specifically in the oncology or rare disease therapeutic areas. However, the FDA is starting to approve more drugs with the help of RWE than before. In a 2019 review of new drug and biologic approvals, 1 in 2 new drug applications (NDAs) and biologic license applications (BLAs) included an RWE study to support safety and/or effectiveness. Moreover, RWE was shown to inform prescribing with 61% of the decisions’ resultant package insert referring to RWE studies and findings. Between 1995 and 1997, 19.4% of the FDA’s approvals came from having one adequate and well-controlled study plus confirmatory evidence, such as RWE. Between 2015 and 2017, the percentage of FDA approvals increased to 47.2% (Purpura 2020; Darrow 2018). In 2019, the FDA approved three products, Pretomanid, Lutathera® (lutetium Lu 177 dotatate), and Zolgensma® (onasemnogene abeparvovec-xioi), in which an externally controlled trial provided the evidence needed to meet the “substantial evidence” standard. Moreover, Bavencio® (avelumab), which is used to treat Merkel cell carcinoma (a rare and lethal form of skin cancer), gained accelerated approval only 3 years after its investigational new drug (IND) filing based on RWE from a retrospective observational study.
However, it is important to note that several gaps exist in RWD. Electronic health records and medical claims data may not capture all of the data elements needed to answer the question of interest and it is challenging to integrate various data sources contributing information about an individual patient. The intent of the FDA’s RWE framework is to address these challenges and limitations.
Review of RWE use in previous regulatory submissions
To gain a better understanding of how RWE has been historically used in FDA submissions, Xcenda conducted a review to contextualize RWE in regulatory decisions. Examples of RWE use were identified from the Drugs@FDA database and the Center for Biologics Evaluation and Research Biological Approval (CBER BLA) list. Within Drugs@FDA, all drugs with submission filters such as efficacy, supplement, type 1 or new molecular entity, or type 2 new active ingredient submission, or any unlabeled submission, were included and reviewed. The approval letter, prescribing information, and medical or multi-disciplinary review was queried for key words indicative of the use of RWE in 1 of the 4 methods outlined by the FDA. From January to July of 2019, 263 drugs from Drugs@FDA and 4 from CBER BLA approvals met the inclusion criteria for key word search. Of the 267 regulatory submissions, 54 unique products, or 20% of drugs queried, were identified to have some component of RWE within their regulatory submission. Overall, there were 17 single-arm trials using RWE as an external control, 6 biosimilars, 13 accelerated approvals, and 9 breakthrough therapies.
While the regulatory submissions were identified during a search completed in 2019, the timing of the original submissions for these drugs dated back to 1979. Searching the prescribing information, medical or multidisciplinary review, and approval letters for even these previously approved therapies found that RWE was being used well before the passing of the 21st Century Cures Act. Our study identified that autoimmune disorders, oncology, and hematology were the top 3 therapeutic areas using RWE, aligning with the FDA framework which states that RWE has primarily been used in cases of oncology and rare diseases (Figure 1).
Figure 1. RWE submissions characterized by therapeutic area

Moreover, observational studies and registry data were the most common RWE submitted (54% and 30%, respectively). However, an increase in the use of modern RWE, such as claims data (7%) and electronic health records (6%), was also observed. Chart reviews (4%) were the least common type of RWE submitted (Figure 2).
Figure 2. Types of RWE submissions
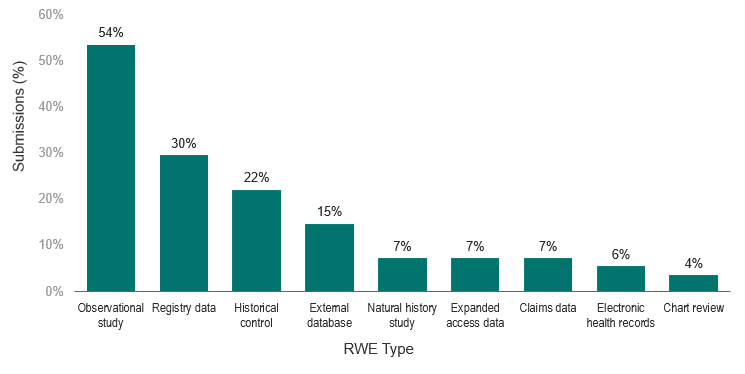
Although observational studies and registry data have commonly been used in post-marketing surveillance, an analysis of the data used in the identified submissions of the review found that these RWE designs are already being used outside of traditional safety evaluations in pregnancy. 76% of the observational studies identified were for non-pregnancy-related studies and registry data were utilized almost equally between pregnancy- and non-pregnancy-related studies (56% and 44%, respectively).
While the FDA RWE framework set a goal to develop future guidance on pragmatic clinical trial design, the review identified examples from past use that can begin to inform design prior to release of further guidance. Real-world baseline characteristics such as allowing the use of prior therapies and continuing concomitant therapies or procedures at baseline were looked upon favorably by the FDA. On the other hand, capped dosages, short durations of therapy, lack of outside factors (eg, alcohol), and excluding discontinued subjects were considered missing pragmatic design features.
Conclusion
The review conducted by Xcenda showed that although RWE has been used prior to the passing of the 21st Century Cures Act, examples of its use are limited by unclear documentation and lack of accessibility. Overall, the utilization of RWE in regulatory submissions appears to be more abundant than previously thought, and there continues to be increased opportunities for its use. However, there is a need for an FDA listing of previous RWE submission metrics such as what is available for other FDA programs. However, until such a database is created, and framework action items are completed, insights to RWE use can be drawn from this categorization of historical submissions.
Integrating RWE into the drug development and approval process has the potential to reduce the time, cost, and patient burden associated with clinical trials, while providing clinically relevant information to all stakeholders. However, challenges such as small sample sizes, data quality, and methodological issues remain. Nevertheless, this review of historical data and a review of the literature indicate that the number of regulatory submissions incorporating RWE to support approval will continue to increase, and a broader scope of RWE across a wider range of indications may be seen in the future. Overall, RWE presents a unique opportunity to accelerate the development of new therapies and to evaluate both the safety and efficacy of these treatments with the FDA RWE framework guiding the correct usage of RWD to develop RWE in order to support regulatory decisions. Given the technical and regulatory challenges related to proper RWE development, it’s imperative that manufacturers seek partners with deep RWE experience to provide strategic guidance.
The article should be referenced as follows:
Patel A, McLaughlin T. Real-world evidence in United States regulatory decisions. HTA Quarterly. Spring 2021. https://www.xcenda.com/insights/htaq-spring-2021-rwe-us-regulatory-decisions
Sources:
- FDA 2020. Real-World Evidence. U.S. Food and Drug Administration. Accessed October 22, 2020.
https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence - Beaulieu-Jones BK, Finlayson SG, Yuan W, et al. Examining the use of real-world evidence in the regulatory process. Clin Pharmacol Ther. 2020;107(4):843-852. doi:10.1002/cpt.1658
- Crown 2020. Unlocking the Promise of Real-World Evidence. ISPOR. Accessed October 22, 2020.
https://www.ispor.org/publications/journals/value-outcomes-spotlight/vos-archives/issue/view/unlocking-the-promise-of-real-world-evidence/unlocking-the-promise-of-real-world-evidence - Darrow JJ, Avorn J, Kesselheim AS. FDA Approval and Regulation of Pharmaceuticals, 1983-2018. JAMA. 2020;323(2):164-176.
https://jamanetwork.com/journals/jama/article-abstract/2758605 - Duh MS. An expanded role for real-world evidence in FDA approvals for drug registration. Analysis Group. 2019. Accessed October 22, 2020.
https://www.analysisgroup.com/Insights/ag-feature/health-care-bulletin/winterspring-2019/expanded-role-real-world-evidence/ - FDA 2019. Demonstrating substantial evidence of effectiveness for human drug and biological products: guidance for industry.
https://www.fda.gov/media/133660/download - FDA 2018. Real-world evidence program. https://www.fda.gov/media/120060/download
- Purpura 2019. Infographic: What role does RWE play in FDA approvals? Accessed October 22, 2020.
https://www.aetion.com/deep-dive/infographic-what-role-does-rwe-play-in-fda-approvals - Purpura 2020. The surge in FDA approvals supported by RWE: A look at three recent FDA decisions. Accessed October 22, 2020.
https://www.aetion.com/post/the-surge-in-fda-approvals-supported-by-rwe-a-look-at-three-recent-fda-decisions


